Recording Neuronal Activity with Light
1. Characteristic of Voltage Sensitive Dye as a Means of Recording Neural Activity
Before going into the main discussion, let us consider the methods of recording brain/neural activity. The substance of neural activity is rapid changes in the membrane potential of neurons, but various recording methods have been developed, some of which reflect membrane potentials of individual neurons, and others of which provide an overall picture of the activity even if the resolution is not high. First, let us briefly discuss neural activity and membrane potentials, which are the subjects of measurement. Neurons are electrically polarized by using differences in ionic concentration as potentials. In the resting state, the inside of the cell is negatively polarized compared to the outside with a potential difference of about 60 mV. This is called the hyperpolarized state, and the potential is called the resting potential. When neurons become active, Na and Ca ions flow into the cell, causing a reversal of the potential, and momentarily (about 1 msec) the potential inside the cell is positively polarized by about 50 mV. This is called depolarization, and this instantaneous potential is called an action potential or spike. When neuronal activity is transmitted from neuron to neuron, a rather slow (about 10 msec) synaptic potential is generated, but its magnitude is about 20 mV.
Fig. 1 summarizes typical methods of recording neural activity and their characteristics. Although all methods are means of recording neural activity, what can be recorded is slightly different. The intracellular recording method (Fig. 1A) measures the membrane potential of neurons by literally inserting an electrode filled with an electrolyte such as KCl into the cell. Therefore, the most accurate recording is possible. If a metallic electrode is placed right next to such a neuron, the local ionic current generated by the spike can be observed. That is the extracellular recording method shown in Fig. 1B. Using an electrode with a sharp tip made of polished tungsten wire or other material, the activity of a single neuron can be recorded. Although the absolute accuracy is lower than that of the intracellular recording, it is possible to observe activity from a large number of neurons by moving the same electrode. It is mainly used to record brain activity in live animals. In contrast, when an electrode with a rounded tip is placed in close contact with the surface of the brain or a slice of the brain, as shown in Fig. 1C, the sum of ionic currents produced by many nearby neurons can be recorded. The advantage of this surface electrode method is that it is position insensitive and easy to handle, whereas the intracellular recording method and extracellular recording methods are very position sensitive and generally difficult to use. However, it has the problem that it does not have temporal and spatial resolution at the cellular level.
The optical method using the voltage sensitive dye shown in Fig. 1D is the subject of this section. As explained above, since neuronal activity is an electrical change, the cell membrane can be dyed with a special dye called voltage sensitive dye to convert the electrical change into an optical change that can be captured in an image.
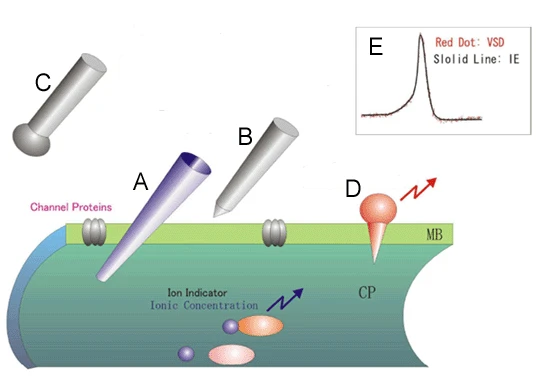
Fig.1 Methods of measuring neural activity
The figure is an enlarged, schematic image of a portion of the neuronal membrane. In the intracellualr recording (A), the neuronal membrane potential is directly recorded by a glass electrode penetrating the neuronal membrane. In contrast, the extracellular recording (B and C) records ionic currents induced by neuronal excitation from outside the cell. In B, an action potential itself is recorded by contacting the neuronal membrane, and a tungsten electrode with a sharp tip is used, while in C, an external electrode with a rounded tip such as silver is used to record the population spike. Voltage sensitive dyes (D) are molecular probes that enter the neuronal membrane and convert membrane potential into fluorescence intensity. The figure shown in E is the action potential of the giant squid nerve axon; the solid black line is the recorded by the intracellualr recording and the red dot is the recorded by the voltage sensitive dye.
The original neural activities are the action potential (100mV/1msec) and synaptic potential (20mV/10msec), and the membrane potential change in which both are mixed can be visualized by the action of the dye. Fig. 1E shows a plot (discrete points) of action potentials at the giant squid nerve axon superimposed on the intracellular recording method (solid line) and changes in light intensity of the voltage sensitive dye. As can be seen, when a single neuron is targeted, the results from two methods are relatively the same. In other words, the greatest advantage of this technique is that what is being recorded is the direct neural activity that is being measured by the intracellular recording method, and there are no difficulties related to electrode insertion, which is generally difficult and requires skill, so that even beginners can easily perform highly reproducible recordings once the setup is in place. Moreover, it is the only method that allows simultaneous observation of more than 1,000 secondary cell-level signals, although the situation varies depending on the optical magnification (focal length of the objective lens) and on the sample. However, this method is not all good. The biggest problem with this method is that the light intensity change due to voltage-sensitive dyes is very small (0.1-1%), resulting in a lot of noise. This problem will be discussed in detail later, but in practical use, it appears as a limitation of various measurement ranges. For example, even with cell-level resolution, it is necessary to reduce noise by adding averages at least 10 times in order to identify and record each individual cell. In addition, when adapting to live animals, the noise caused by vibration due to heartbeats and respiration is a major problem.
As described above, optical recording using voltage-sensitive dyes, like the electrode method, is a means of recording neural activity itself (electrical signals) in real time, and it is a method that realizes multi-point measurement, which is impossible with the electrode method. Fig. 2 shows the range of application. By changing the magnification of the optical system, the range of application can be extended from the organ level to the subcellular level. Probably the most popular method at present is the imaging of neural networks with 1 to 3x magnification, which allows us to easily record unknown neural circuits. For example, it is expected to be a powerful tool for research to observe the unknown brain of a genetically defective model animal (knockout mouse, etc.). Another simple and difficult research subject is how electrical signals are generated and propagated inside a single neuron under higher magnification (10x or more), but it is important for elucidating the function of functional molecules and other functions of the neuron. On the other hand, research using animals whose brain surfaces are exposed through a lens with a magnification of less than 1x has yielded remarkable results, and this technique is the sole field of application for elucidating the dynamic representation of information inside the brain. In addition to cranial nerves, this technique has begun to be applied as an effective means of observing electrical excitation in cardiac muscle and adrenal glands.
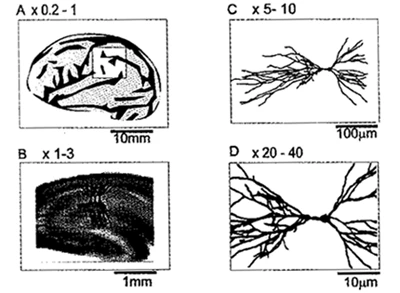
Fig. 2 Applicable range of optical recording using voltage sensitive dye
The figure contrasts the magnification of the optical system with the object of imaging. With a reduced magnification lens (A, 0.2 to 1x), the entire brain or a part of the brain is observed; with a low magnification lens (B, 1 to 3x), the neural network is observed; and with a higher magnification, a single cell (C) or even a synapse (D) can be resolved.
2. Problems in Optical Recording - Noise and Signal
As mentioned in the previous section, this technique is very useful and not very difficult. However, sufficient imaging results cannot be obtained without a great deal of knowledge and know-how. For example, if one wants to reduce noise, one should naturally make the light brighter. However, intense illumination induces dye denaturation and sample damage, resulting in inaccurate imaging. In order to obtain good results with voltage-sensitive dyes, it is necessary to address the issues of how to minimize illumination, how to minimize staining, and yet still capture a bright image. Naturally, this requires a great deal of effort and compromise. The relationship between noise, signal, and light intensity is shown in Fig. 3. The horizontal axis is the total amount of fluorescence emitted by the dye, and the vertical axis is the component of change in fluorescence, both on a logarithmic axis. The numbers are the number of photons of light exposed to one pixel (about 35 µm square) of the sensor we are using; the two 45-degree solid black lines represent the signal of a typical voltage sensitive dye (Di-4-ANEPPS) changing due to neuronal activity, with the top line at 1% and the bottom line at 0.1% line. There are variations in the magnitude of the signal depending on the sample, but in most cases, values such as 0.2% or 0.6% fall between these two lines. The noise of the imaging device itself is generally independent of the light level, so the line will be horizontal, as shown by the red dotted line. It is almost impossible to image areas darker than this, but in many cases it is easy to go beyond this line. In practice, the most problematic noise is the photon shot noise, which increases with the square root of the light intensity, indicated by the red dashed line. Photon shot noise is noise based on the particle nature of light (photons) and is physically inescapable. Above this line, the signal exceeds the noise and the signal-to-noise ratio is greater than 1. The area indicated by * in the figure is the area where the signal-to-noise ratio is practically 1. Naturally, a brighter area to the right of this line can be measured with a better signal-to-noise ratio.
The problem, however, is that no such bright optical system (fluorescence microscope) exists. In order to make it brighter, the illumination must be stronger, but this causes more damage to the sample. The amount of fluorescence that can be obtained with ordinary fluorescence microscopes is in the range indicated by the arrow "easy" in the lower right corner of Fig. 3, and the amount of fluorescence indicated by the arrow "high NA" can be achieved when a lens with a particularly high aperture is devised. There are currently no brighter fluorescent microscopes than that, and the only way to make them brighter is to increase the intensity of the light source. Usually, a 100-150W halogen bulb is used as a light source, but even if it is changed to a 500W-class light source, the brightness is improved by a factor of 3 to 5, and the improvement in S/N is at most a factor of 2 since it is the square root. However, since sample damage deteriorates linearly or more than that with respect to illumination, the S/N improvement is empirically about 10 times worse. In other words, a 2-fold improvement is obtained at the expense of a 10-fold improvement. Of course, if the purpose of an experiment is to achieve a high signal-to-noise ratio just once, then an intense light source should be provided without hesitation. However, it is also true that in many cases, experiments are constructed on the premise of stable measurements over long periods of time.
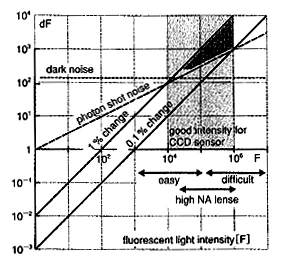
Fig.3 Examination diagram of signal and noise of optical measurement
The horizontal axis shows the amount of fluorescence that enters into one pixel of the sensor, and the vertical axis is the component of the change. 2 solid lines are the signal and the dashed line is the noise. In the region where the signal exceeds the noise (marked with *), it is possible to observe S/N = 1 or so.
As described above, as long as the fact that the change in voltage-sensitive dyes is less than 1% remains unchanged, we must admit that the signal-to-noise ratio cannot be improved. To overcome this problem, efforts must be made to (1) use a bright lens system, (2) find the staining method with the highest signal and the least damage, and (3) remove noise by data processing. Of these, (2) is largely a matter of the experimenter's own efforts and must be found experimentally (see the following column), while (1) and (3) can be achieved through the development of equipment. The most important thing, then, is to clarify what it is that one wants to see and to thoroughly examine the measurement protocol. For example, one may want to observe asynchronous phenomena and therefore cannot use averaging. However, it is very worthwhile to consider whether it is possible to simultaneously record another measurement (e.g., an extracellular electrode) that produces some synchronous signal and then apply synchronization to that signal. If so, the signal-to-noise ratio can be greatly improved by averaging.
We will present staining methods that have been empirically discovered in our laboratory to be effective in rat and mouse cerebral slice specimens.
| Empirically found effective staining methods | |
|---|---|
| (1) | Dissolve 1 ml of alcohol directly in a bottle of Di-4-ANEPPS (5 mg) and dissolve
well. (The catalog says methanol, but ethanol dissolves better.) |
| (2) | Add 0.5 ml of Cremophor-EL 10% aqueous solution and dissolve well to make a stock solution. It can be stored in the refrigerator for 2 to 3 months. |
| (3) | Dissolve 35 µm of the above stock solution in 0.5 ml fetal bovine serum and 0.5 ml ACSF to make a staining solution. The final concentration is approximately 0.1 mg/ml. |
| (4) | The slices are made in a sealed staining container that can be kept like an interface chamber, and a small amount of the above staining solution is dripped onto the surface of the slices and allowed to stand for about 20 minutes. The temperature is room temperature, and the ACSF is kept oxygenated and humid by publing 5% carbon dioxide and oxygen gas inside the container. |
| (5) | The slices are taken to the observation chamber (submersion type, placed under the microscope) and incubated for about 30 minutes before observation. This method allows stable observation for about 4 hours. |
3. High NA Optics and High-Speed Camera System
The light collection efficiency of an optical system depends mainly on the numerical aperture (NA) of the objective lens. When the number of apertures is doubled, the condensing lens system is 4 times brighter and the illumination system is 4 times brighter, for a total of 16 times brighter images. In other words, brightness is proportional to the fourth power of the numerical aperture. A lens with a high numerical aperture means a large aperture and a short focal length. Therefore, it is brighter because it captures light spreading from the subject at a wider angle. In the case of commercially available microscope lenses, those with apertures of about 0.7 at 40x, 0.3 at 20x, and 0.1 at 10x are commonly used. However, as shown in Fig.2, the most common applications, such as observation of neural circuits, require optics with a low magnification of 1 to 3x. The 2x lenses sold by most microscope manufacturers have an aperture of about 0.05 and are extremely dark. Therefore, we have developed a special optical system as shown in Fig.4. The objective lens is a Nikon single-lens reflex lens (f/1.2, 50mm standard lens) disassembled and reassembled in a case with extra parts (aperture and focusing mechanism) removed. The aperture of this lens is approximately 0.7. Compared to commercially available microscope lenses, this lens produces images 200 times brighter.
This custom-made microscope has a so-called infinity design with two lenses of high numerical aperture. A nearly parallel beam of light passes through the body where the dichroic mirror is placed, passing through the projection lens and forming an image on the CCD sensor. The projection lens is an 80 mm stereo microscope lens made by Leica. The total magnification is approximately 1.6x, and an absorption filter is placed to pass only longer wavelengths than 590nm.
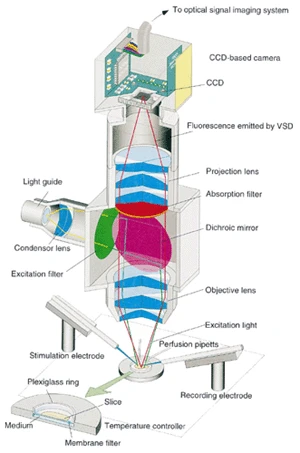
Fig.4 The optical system is a custom-made coaxial epifluorescence microscope assembled around an objective lens with a large numerical aperture. The yellow line shows the illumination light and the red line shows the fluorescence light. A dichroic mirror is used to separate the two. The reason for the brightness of this microscope is the objective lens, which was made by dismantling a camera lens (Nikon F1.2, 50mm). A slice of brain, the sample, is placed under the objective lens, and electrodes, etc., are set in place. The total magnification is approximately 1.6x.
The illumination system uses a fiber lighting device (Moritex, MHF-G150) with a commercially available 150W halogen bulb. To match the wavelength characteristics of the dye, the illumination is green light (530 nm) through an interference filter. A dichroic mirror reflects more than 90% of the 530 nm light, which is dropped onto the sample. Since the light between 590 and 650 nm is the voltage-sensitive component of the fluorescence at the sample, to effectively cut this out, the dichroic mirror transmits more than 90% of the light between 590 and 650 nm, followed by an absorption filter that passes only longer wavelengths than 590 nm to block stray light.
The camera system is a high-speed CCD imaging system developed by our laboratory and Brain Vision. The CCD sensor used in this camera is a SONY ICX076, a monochrome CCD image sensor for normal use. Normally, this component should be operated at a clock speed of about 7 MHz, but we forced it to operate at 50 MHz. In addition, the CCD transfer clock in the vertical direction (V-direction) is given four times in succession, and the number of vertical scan lines is summarized to 60 lines to achieve high-speed operation. As a result, the entire image can be read out in approximately 700 µsec. Considering that neural activity is about 1 msec, 700 μsec is not necessarily a sufficient speed, but it is a speed that generally meets the requirements. The CCD sensor is 1/5 inch in size, with an effective photosensitive area of approximately 3 mm x 2 mm. Images captured by the CCD sensor are processed and stored digitally through a high-speed ADC. The video data is temporarily stored in memory and transferred to the host computer via the PCI bus immediately after measurement.
4. Imaging Result
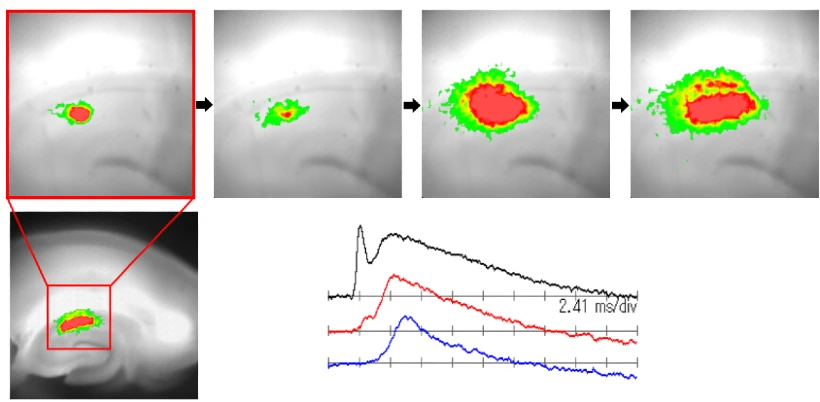
Using the camera system and optics described in the previous section, samples obtained from live brain slices were stained with Di-4-ANEPPS dye, and the result of imaging with small electrical stimuli are shown in Fig.5. Fig.5A shows the imaging area and the arrangement of electrodes and other components for the excised rat brain. The imaging area is the part of the hippocampus called CA1. Stimulation electrode is placed in the Schaffer collateral, which is the main input fiber in the CA1 region, and are electrically stimulated with short pulses. Stimulation excites the pyramidal cells of CA1 one after another through the Schaffer collateral. This can be clearly seen in Fig.5C. The images were originally caputured at 0.75 msec/frame, but for the convenience of the figure, images are shown every other picture, i.e., every 1.5 msec. Since the change in voltage-sensitive dye due to neural excitation is originally about 0.3%, only the change component is emphasized about 300 times by computer processing and superimposed on the image with a red color. The darker red areas are the more depolarized areas. Thus, the greatest advantage of this method is the ability to image the membrane potential in two dimensions in real time. Furthermore, Fig.5B shows a time-dependent analysis of neural activity at selected points in the slice (in this case, four points corresponding to each location of the CA1 pyramidal cells). This is just about equivalent to stabbing multiple internal electrodes, but in reality, the data is read from each pixel recorded as a video image at the same time. In other words, we can think of an internal electrode as being present at any point on this image. In Fig.5B, the solid line with little noise shown below the four traces is the action potential recorded at the internal electrode, and the small red circle is the result of the optical imaging at almost the same location. Thus, it can be seen that almost identical waveforms are obtained. However, upon closer inspection, the slow component appears slightly larger in the optical measurement compared to the action potential peak. This is because (1) the action potential observed with light is an average of several cells, so the fast action potential is less synchronized and observed smaller than it actually is, and (2) the amount of voltage-sensitive dye measured is proportional to membrane density as well as membrane potential. In this sample, the density of neuronal membranes is high in dendrites and low in cell bodies, so that the synaptic potential component is accentuated when the dendrite component is mixed in, for two possible reasons. In studies using voltage-sensitive dyes, differences in staining by tissue and the density of active membranes should be considered. It should be noted that some dyes (e.g., RH-155) are known to be taken up by glia more often than neurons, so that the relatively slow polarization of glia can be mixed into the data. The Di-4-ANEPPS used for this measurement shows almost all of the fluorescence changes dependent on neuronal activity, with little glial activity visible. For completely new samples, the compatibility with the dye used and other factors should be investigated before proceeding to detailed experiments.
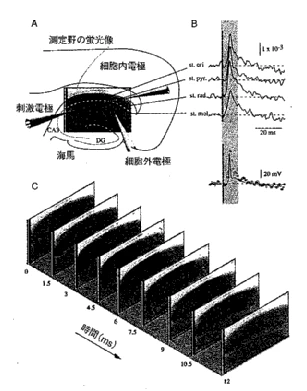
Fig.5 Hippocampus was removed from rat brain, sliced into approximately 400 μm thick, and stained with Di4-ANEPPS. The imaging area is CA1 (see Fig.A), and the stimulus is a current in the Schaeffer collateral projecting from CA3 to CA1. Shown in Fig.B is a plot of the amount of change in fluorescence of pixels in each of the areas indicated by lines in Fig.A versus time. Shown in the bottom panel of Fig.B is the membrane potential measured at the internal electrode (solid line) overlaid with a photometric point (red circle) at approximately the same location. Fig.C is a sequence of images of time (in msec) at the bottom left of each figure. The data in this figure has been averaged 8 times and filtered for S/N improvement.
Conclusion
Measuring neural excitatory activity with voltage-sensitive dyes is not a difficult method once you get a few points. In addition, this technique is historically old, having already been developed 20 years ago. However, it is still generally considered a difficult technology. We reflect that the reasons for this are the difficulty in obtaining a suitable camera system for the measurement and the fact that the steady accumulation of data to realize a systematic and stable measurement has come later, and we have been looking for flashy results. One direction of our research is to perfect optical imaging techniques that anyone can use. Synchronously, research institutes around the world are also making progress in introducing GFP (green fluorescent protein from jellyfish) into channel proteins at the genetic level. In the near future, there is a possibility that experimental animals will be born with brains that change fluorescence according to membrane potential without being electrically decorated. If this happens, the use of this technology will spread rapidly.
Now, so far, there are no examples of implementation in medical diagnosis on human subjects. The main reason for this is that, unlike MRI, which is a completely non-invasive technique, the area to be diagnosed must be visible. Also, although the dye is less toxic, it is not safe. However, it is heard that the application of MRI is being considered as an adjunctive means to improve accuracy in cardiac and brain surgery, mainly at medical universities in the United States. In the years to come, it may develop into a method that can save many lives. I would like to conclude this section by expressing my hope for the arrival of such a day.
(Translated using Neural Machine Translation)
(Source: Kyoritsu Shuppan, "Medical Diagnosis by Light" (published March 30, 2001))